exSEEK
exSEEK is an integrated computational framework to discover and evaluate exRNA biomarkers for liquid biopsy.
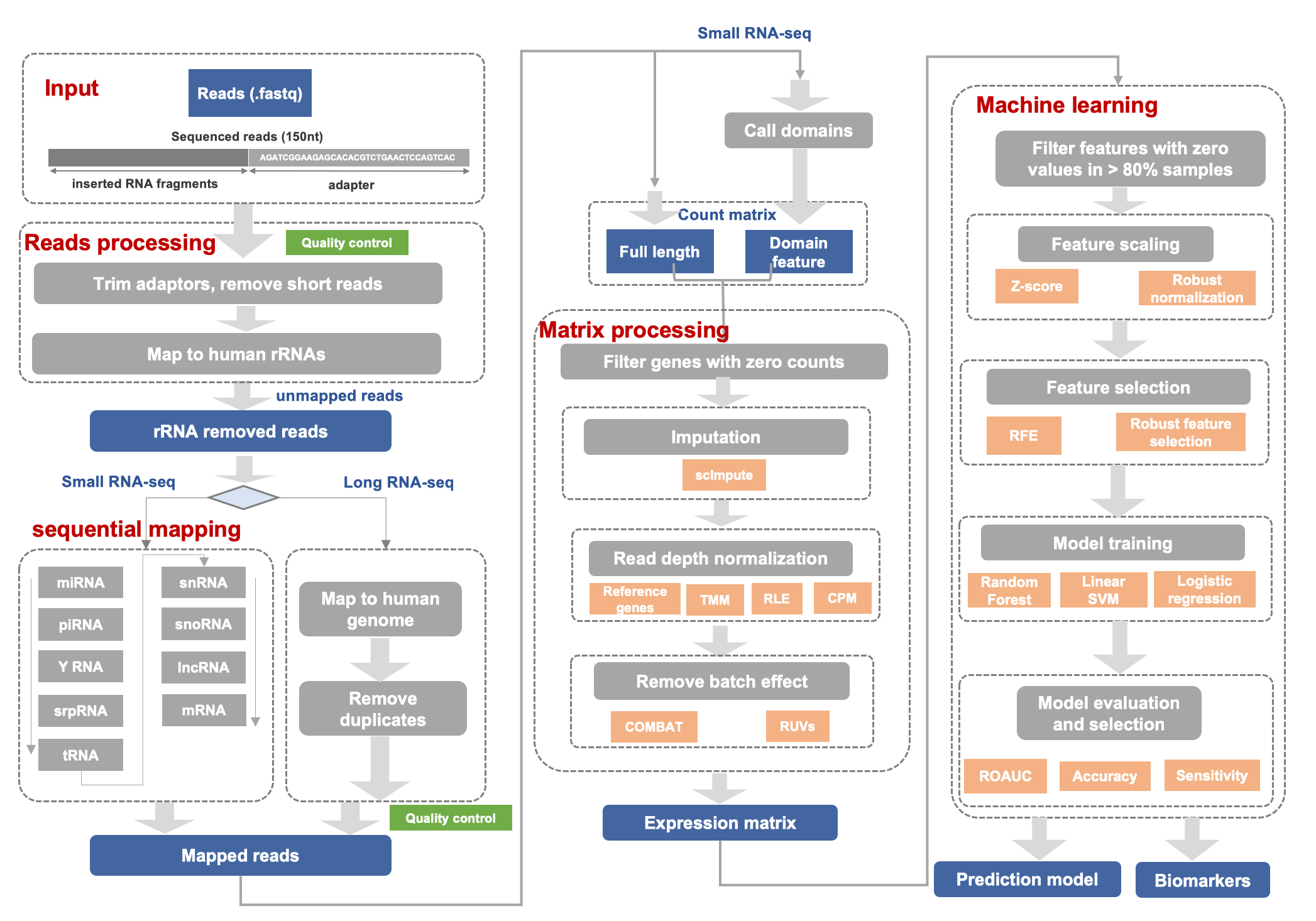
The exSEEK framework consists of:
-
Pre_processing:
- Building index with various types of genomes and annotations. [
exseek build-index] - Quality control and removing adaptors. [
exseek quality_control] [exseek cutadapt] [exseek quality_control_clean] - Sequential mapping for small/long RNA-seq. [
exseek mapping]
- Building index with various types of genomes and annotations. [
-
Main function:
- Peak calling for recurring fragments of long RNAs. [
exseek bigwig] [exseek call_domains] - Counting expression matrix. [
exseek count_matrix] - Normalization and batch removal. [
exseek normalization] - Feature selection and classification. [
exseek feature_selection] - Biomarker evaluation. [
exseek feature_selection]
- Peak calling for recurring fragments of long RNAs. [
Table of Contents:
Installation
For easy installation, you can use the exSEEK image of docker with all dependencies installed:
docker pull ltbyshi/exseek
All required software and packages are already installed in docker, so there are no more requirements. To test the installation and get information about the command-line interface of exSEEK, you can execute:
docker run --rm -it -v $PWD:/workspace -w /workspace ltbyshi/exseek exseek.py -h
The -v flag mounts the current working directory $PWD into the /workspace in docker image, so you can easily check the output files in /workspace directory after exiting docker.
You can create a bash script named exseek and set the script executable:
#! /bin/bash
docker run --rm -it -v $PWD:/workspace -w /workspace ltbyshi/exseek exseek.py "$@"
After adding the file to one of the directories in the $PATH variable, you can simply run: exseek.
A helper message is shown:
usage: exseek.py [-h] --dataset DATASET [--config-dir CONFIG_DIR] [--cluster]
[--cluster-config CLUSTER_CONFIG]
[--cluster-command CLUSTER_COMMAND] [--singularity]
{build_index,quality_control,cutadapt,quality_control_clean,mapping,
call_domains,count_matrix,normalization,feature_selection}
exseek main program
positional arguments:
{build_index,quality_control,cutadapt,quality_control_clean,mapping,
call_domains,count_matrix,normalization,feature_selection}
optional arguments:
-h, --help show this help message and exit
--dataset DATASET, -d DATASET dataset name
--config-dir CONFIG_DIR, -c CONFIG_DIR directory for configuration files
--cluster submit to cluster
--cluster-config CLUSTER_CONFIG cluster configuration file ({config_dir}/cluster.yaml by default)
--cluster-command CLUSTER_COMMAND command for submitting job to cluster (default read from
{config_dir}/cluster_command.txt
--singularity use singularity
The basic usage of exSEEK is:
exseek ${step_name} -d ${dataset}
Note:
${step_name}is one of the step listed in ‘positional arguments’.${dataset}is the name of your dataset that should match the prefix of your configuration file described in the following section.
Usage
You can use the provided example_data to run exSEEK:
cp /apps/example_data /workspace
The example_data folder has the following structure:
example_data/
├── config
| ├── example.yaml
| ├── default_config.yaml
│ └── machine_learning.yaml
├── data
│ └── example
| ├── fastq
│ ├── batch_info.txt
│ ├── compare_groups.yaml
│ ├── sample_classes.txt
│ └── sample_ids.txt
└── output
└── example
└── ...
Note:
config/example.yaml: configuration file with frequently adjusted parameters, such as file paths and mapping parameters.config/default_config.yaml: configuration file with additional detailed parameters for each step. The default file is not supposed to be changed. If you want to adjust parameters contained in this file, it is recommended to add your adjusted parameters inconfig/example.yaml.config/machine_learning.yaml: configuration file with parameters used for feature selection and classification steps. It is recommended to add your adjusted parameters inconfig/example.yamlif you want to adjust parameters contained in this file.data/example/batch_info.txt: table of batch information.data/example/compare_groups.yaml: table for definition of positive and negative samples.data/example/sample_classes.txt: table of sample labels.output/example/: output folder.
Index preparing
exSEEK docker contains a variety of commonly used genomes and annotations. Besides of RNA types extracted from GENCODE V27, exSEEK can also analyze rRNA from NCBI refSeq 109, miRNA from miRBase, piRNA from piRNABank, circRNA from circBase, lncRNA and TUCP from mitranscriptome, repeats from UCSC Genome Browser (rmsk) and promoter and enhancer from ChromHMM tracks. You can download these .fa, .gtf and .bed files here and build index with these genomes and annotations.
For mapping small RNA-seq, the index of each transcript type can be built with bowtie2, and the index used for mapping long RNA-seq can be built with STAR. You can get these index by executing:
exseek build_index -d example
It might take hours to generate the index. It is recommended to specify the number of threads in config/example.yaml file by adding threads: N, or you can simply add -j N parameter in the exseek command.
The output folder is genome/hg38/index/.
The detailed information for each transcript type is in genome/hg38/transcript_table/ directory.
The summary for transcript types is listed below:
| RNA type | Number of transcripts |
| :— | :— |
| rRNA | 37 |
| mature_miRNA | 2656 |
| miRNA | 1917 |
| piRNA | 23410 |
| snoRNA | 943 |
| snRNA | 1900 |
| srpRNA | 680 |
| tRNA | 649 |
| mRNA | 19836 |
| lncRNA | 15778 |
| TUCP | 3730 |
| Y_RNA | 756 |
| univec | 6093 |
| spikein_long | 92 |
| spikein_small | 52 |
Small RNA-seq mapping
Quality control (before adaptor removal)
You can check reads quality with FastQC by running:
exseek quality_control -d example
Note:
- The detailed results for each sample are in folder
output/example/fastqc/.- You can quickly check the summary results for all samples with the
fastqc.txtfile inoutput/example/summary/fastqc_data/multiqc_fastqc.txt.
Remove adapter
exSEEK removes reads adaptor with cutadapt software. You can change the adaptor sequences in config/example.yaml file.
exseek cutadapt -d example
Note:
- You can check the additional parameters for cutadapt in
config/default_config.yamlfile.- You can check the adaptor revmoval summary with
output/example/summary/cutadapt.txtfile.
Quality control (after adapter removal)
exseek quality_control_clean -d example
Note:
- The detailed results for each sample are in folder
output/example/fastqc_clean/.- You can quickly check the summary results for all samples with the
fastqc.txtfile inoutput/example/summary/fastqc_clean_data/multiqc_fastqc.txt.
Update sequential mapping order
exSEEK allows user-defined sequential mapping, which is particularly useful for small RNA-seq samples because short RNA reads are more likely to be mapped to multiple locations. The default mapping order is set as rna_types variable in config/default_config.yaml:
rna_types: [univec, rRNA, lncRNA, mature_miRNA, miRNA, mRNA,
piRNA, snoRNA, snRNA, srpRNA, tRNA, tucpRNA, Y_RNA]
You can change the mapping order based on the confidence of each RNA type in your samples by adding a rna_types variable in config/example.yaml. For example, you can add spike-in sequences as the first RNA type:
rna_types: [spikein_small, univec, rRNA, lncRNA, mature_miRNA, miRNA,
mRNA, piRNA, snoRNA, snRNA, srpRNA, tRNA, tucpRNA, Y_RNA]
Add new reference sequence
If a new RNA type is added, you should also add a sequence file in FASTA format: ${genome_dir}/fasta/${rna_type}.fa. Then build a FASTA index (${genome_dir}/fasta/${rna_type}.fa.fai):
samtools faidx ${genome_dir}/fasta/${rna_type}.fa
Then build a bowtie2 index (${genome_dir}/index/bowtie2/${rna_type}):
bowtie2-build ${genome_dir}/fasta/${rna_type}.fa ${genome_dir}/index/bowtie2/${rna_type}
Mapping
exSEEK provides bowtie2 for mapping small RNA-seq. You can specify the paired_end parameter as false or true in config/example.yaml. It is recommended to specify the number of threads in config/example.yaml file by adding threads_mapping: N, or you can simply add -j N parameter in the exseek command. The other parameters for mapping can be found in config/default_config.yaml.
exseek mapping -d example
Note:
- Make sure that the parameter
small_rnaisTrueinconfig/example.yaml.- The output folder
output/example/gbamcontains genome bam files.- The output folder
output/example/tbamcontains transcriptome bam files for all types of RNA.- The output folders
output/example/stats/mapped_read_length*/contain the summary of read length distribution for each RNA type.- The output file
output/example/summary/read_counts.txtis the summary of read counts mapped to each RNA type for all samples.
Peak calling
exSEEK provides local maximum-based peak calling methods for identifying recurring fragments (which are recurrently detected among samples,defined as domains) of long exRNAs (such as mRNA, srpRNA, and lncRNA). These called domains can be combined into the expression matrix and serve as potential biomarker candidates.
exseek bigwig -d example
exseek call_domains -d example
Notes:
- Domain calling parameters in
config/default_config.yaml:bin_size: 20: size of bins for calculating read coverage.cov_threshold: 0.05: The proportion of samples that have the called peak. Peaks with cov_threshold higher than 0.05 are defined as domains.
- Output files:
output/example/domains_localmax_recurrence/recurrence.bedcontains all recurring peaks (domains).output/example/domains_localmax/domains.bedcontains filtered (domains shorter than 10nt are filtered out) and merged domains.
The recurrence.bed file looks like this:
| Transcript ID | TransStart | TransEnd | X | Frequency | Strand |
| :— | :— | :— | :— | :— | :— |
| ENST00000365118.2 | 0 | 30 | X | 8 | + |
| ENST00000365223.1 | 0 | 61 | X | 12 | + |
| ENST00000365436.1 | 69 | 92 | X | 2 | + |
| ENST00000366365.2 | 236 | 261 | X | 1 | + |
The domains.bed file looks like this:
| Transcript ID | TransStart | TransEnd | filtered_merged Peak_ID | Weighted_ave_frequency | Strand |
| :— | :— | :— | :— | :— | :— |
| ENST00000006015.3 | 1506 | 1523 | peak_1 | 14.2353 | + |
| ENST00000006015.3 | 1971 |1986 | peak_2 | 10 | + |
| ENST00000008938.4 | 20 | 35 | peak_3 | 7 | + |
| ENST00000025301.3 | 8580 | 8597 | peak_4 | 37.2353 | + |
| ENST00000192788.5 | 2649 | 2665 | peak_5 | 72.5625 | + |
Long RNA-seq mapping
The methods for long RNA-seq mapping are very similar to Small RNA-seq mapping. You can use the above command lines for long RNA-seq by setting small_rna to False in file config/example.yaml. It is recommended to specify the number of threads in config/example.yaml file by adding threads_mapping: N, or you can simply add -j N parameter in the exseek command. There is no peak calling step for long RNA-seq datasets because recurring fragment (domain) is not a distinctive feature of extracellular long RNA-seq datasets.
Counting expression matrix
exSEEK use featureCounts for counting expression matrix.
exseek.py count_matrix -d example
Notes:
- For small RNA-seq, the ouput folder
output/example/count_matrix/contains 4 types of expression matrix:Name Transcript type transcript.txt all full_length transcripts transcript_mirna.txt only miRNA long_fragments.txt recurring peaks (domain) mirna_and_long_fragments.txt miRNA and recurring peaks (domain) - For long RNA-seq, the ouput folder
output/example/count_matrix/contains 2 types of expression matrix:Name Transcript type featurecounts.txt genome_long_rna circRNA.txt circRNA
Normalization and batch removal
exSEEK supports 5 normalization methods and 4 batch removal methods in config/example.yaml:
normalization_method: ["TMM", "RLE", "CPM", "CPM_top", "null"]
batch_removal_method: ["null", "ComBat", "limma", "RUV", "null"]
count_method: [transcript, transcript_mirna, long_fragments, mirna_and_long_fragments, featurecounts(for long RNA-seq)]
batch_index: 1
You can get the normalized expression matrix generated by any combinations of normalization and batch removal methods by executing:
exseek normalization -d example
Notes:
- When the method name is set to
null, the step is skipped.- You can specify the expression matrix type to be normalized via the
count_methodvariable.batch_indexis the column index ofdata/example/batch_info.txtto be used for ComBat batch removal.- The name pattern of output files in folder
output/example/matrix_processingis:filter.null.Norm_${normalization_method}.Batch_${batch_removal_method}_${batch_index}.${count_method}.txt.
You can choose the best combination methods based on the UCA score and then mKNN score, which is summarized in folder: output/example/select_preprocess_method/uca_score/ and output/example/select_preprocess_method/knn_score.
The UCA metric quantifies the separation of samples from different biological groups, while the mKNN metric measures the uniformity of the distribution of samples from different batches. For a perfectly corrected expression matrix, both the UCA score and the mKNN score approach 1.
The UCA score files look like this: | preprocess_method | uca_score | | :— | :— | | filter.null.Norm_CPM_top.Batch_limma_1 | 0.578 | | filter.null.Norm_CPM.Batch_limma_1 | 0.563 | | filter.null.Norm_CPM_top.Batch_ComBat_1 | 0.563 | | filter.null.Norm_CPM_top.Batch_RUV_1 | 0.564 |
And the mKNN score files look like this: | preprocess_method | knn_score | | :— | :— | | filter.null.Norm_CPM_top.Batch_limma_1 | 0.940 | | filter.null.Norm_CPM.Batch_limma_1 | 0.936 | | filter.null.Norm_CPM_top.Batch_ComBat_1 | 0.936 | | filter.null.Norm_CPM_top.Batch_RUV_1 | 0.927 |
Alternatively, you can simply get the best-performance combination method (the highest averaged UCA and mKNN score) listed in output/example/select_preprocess_method/combined_score/${count_method}/selected_methods.txt.
After deciding the most proper combination of normalization and batch removal methods, you can specify the exact normalization and batch removal method by adjusting normalization_method and batch_removal_method parameters in config/sample.yaml, and the generated matrix will be used for the next step.
Feature selection and biomarker evaluation
This step identifies and evaluates exRNA biomarker panels selected by various combinations of feature selection methods and machine learning classifiers.
exSEEK supported feature selection and classification methods:
selector: [DiffExp_TTest, RandomForest, LogRegL1, LogRegL2, SIS, ReliefF, SURF, MultiSURF]
classifier: [LogRegL2, RandomForest, RBFSVM, DecisionTree, MLP]
You can evaluate all combinations of feature selection and classification methods based on the cross-validation results by running:
exseek feature_selection -d example
Note:
- You can adjust the maximum number of selected features
n_features_to_selectinconfig/example.yaml.- You can setup the comparison groups for classification in
data/config/compare_groups.yaml.- The detailed parameters of machine learning can be found in
config/default_congfig.yaml.- The cross-validation results and trained models for individual combinations are in this directory:
output/example/cross_validation/filter.null.Norm_${normalization_method}.Batch_${batch_removal_method}_${batch_index}.${count_method}/${compare_group}/${classifier}.${n_select}.${selector}.${fold_change_filter_direction}.- Selected features (biomarker panels) for each model can be found in
features.txtin the above-mentioned directory.
Three summary files will be generated in this step:
output/example/summary/cross_validation/metrics.test.txt
output/example/summary/cross_validation/metrics.train.txt
output/sxample/summary/cross_validation/feature_stability.txt
You can choose the most proper combination and its identified features (biomarker panel) base on ROC_AUC and feature stability score summarized in the above three files.
The metrics.*.txt file looks like:
| classifier | n_features | selector | fold_change_direction | compare_group | filter_method | imputation | normalization | batch_removal | count_method | preprocess_method | split | accuracy | average_precision | f1_score | precision | recall | roc_auc |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LogRegL2 | 5 | MaxFeatures_RandomForest | any | Normal-HCC | filter | null | Norm_RLE | Batch_limma_1 | mirna_and_domains_rna | filter.null.Norm_RLE.Batch_limma_1 | 1 | 0.928 | 0.916 | 0.800 | 1.000 | 0.666 | 0.969 |
| LogRegL2 | 5 | DiffExp_TTest | any | Normal-HCC | filter | null | Norm_RLE | Batch_limma_1 | mirna_and_domains_rna | filter.null.Norm_RLE.Batch_limma_10 | 1 | 0.928 | 0.743 | 0.800 | 1.000 | 0.666 | 0.696 |
The feature_stability.txt file looks like:
| classifier | n_features | selector | fold_change_direction | compare_group | filter_method | imputation | normalization | batch_removal | count_method | preprocess_method | feature_stability |
|---|---|---|---|---|---|---|---|---|---|---|---|
| LogRegL2 | 5 | DiffExp_TTest | any | Normal-HCC | filter | null | Norm_RLE | Batch_limma_1 | mirna_and_domains_rna | filter.null.Norm_RLE.Batch_limma_1 | 0.450 |
| RBFSVM | 5 | DiffExp_TTest | any | Normal-stage_A | filter | null | Norm_RLE | Batch_limma_1 | mirna_and_domains_rna | filter.null.Norm_RLE.Batch_limma_1 | 0.473 |
Copyright and License Information
Copyright (C) 2019 Tsinghua University, Beijing, China
This program is licensed with commercial restriction use license. Please see the LICENSE file for details.