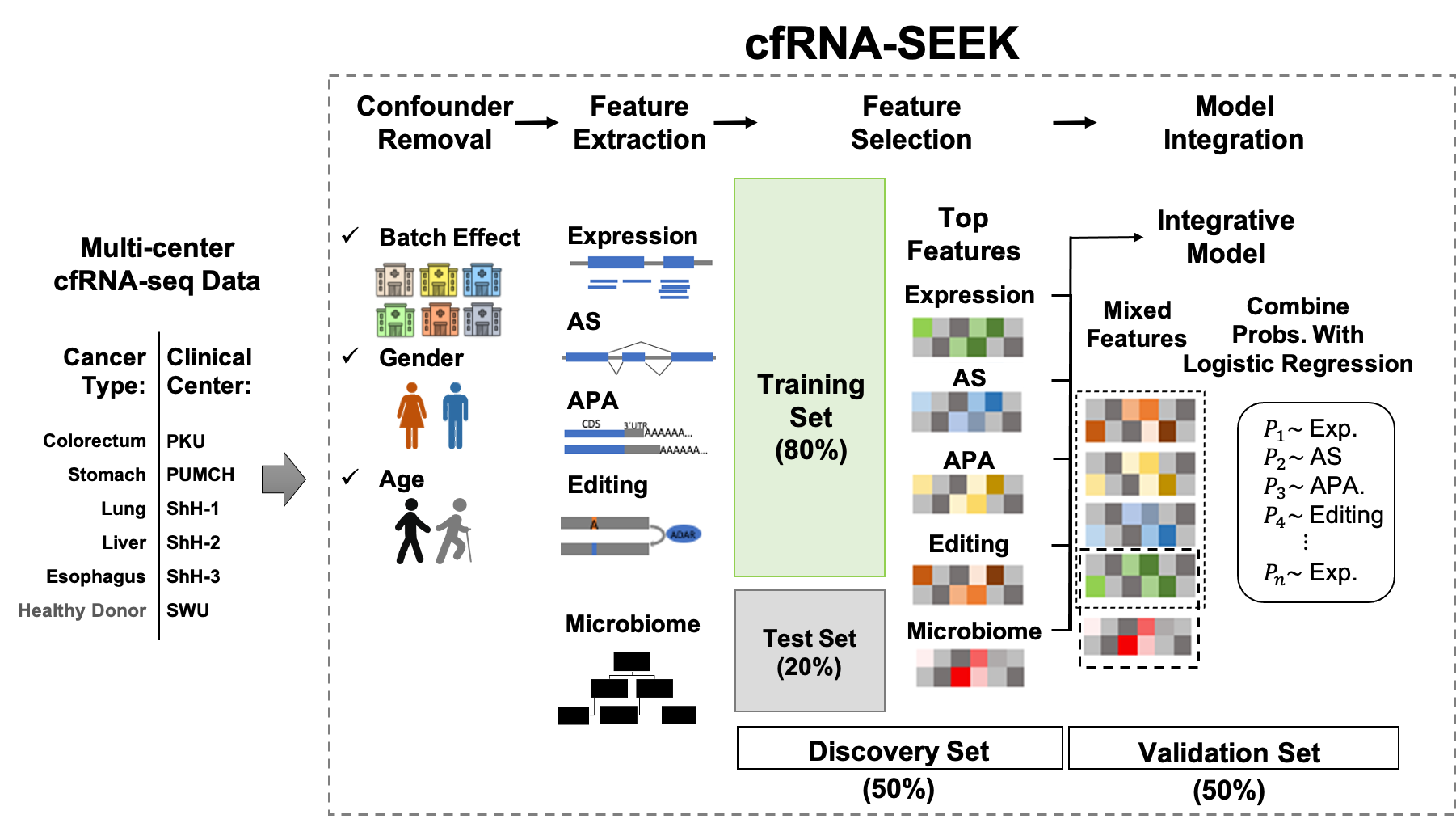
cfRNA-SEEK
Website hosted by github page: https://lulab.github.io/cfRNA-SEEK
Scripts for cfRNA Sequencing Data Analysis
This repo contains scripts used in cfRNA-SEEK, a pipeline for identification of cancer relevant features from cfRNA sequencing (cfRNA-seq) data generated by SMART-Total protocol. Scripts for small cfRNA-seq data processing are also available. We also provide snakemake files to tidy up the scripts.
- SMART-total cfRNA-seq Reads Processing
To quantify different RNA variations for human genome, as well as the abundance of different microbial taxa, see analysis-step-smart-total-libraries.md.
- Analysis of unmapped reads
To analysis unmapped reads, see microbe-analysis.md
- Feature Selection
To nomalize data matrix, remove batch effect, identify cancer relevant features and evaluate the classification performance, see classification.md.
- Small cfRNA-seq Reads Processing
To analyze small cfRNA-seq data, see analysis-step-small-RNA-libraries.md.
-
Dependency
program version purpose cutadapt 2.3 Trim adaptor and low quality sequence STAR STAR-2.5.4a Reads alignment bowtie2 2.3.5 Reads alignment MarkDuplicates (picard toolkit) 2.20.0 Duplication removal samtools 1.9 Manipulation of bam file, quanltification of editing level bedtools 2.28.0 Assign mapped reads to different genomic regions rMATs 4.0.2 Alternative splicing analysis featureCounts 1.6.2 Quantify gene expression DaPar 0.91 APA analysis RNAeditor 1.0 Identify RNA editing sites edgeR 3.24.3 Normalization and differential expression analysis RUVSeq 1.6.1 Normalization use control genes scipy 1.4.1 Perform ranksum test sklearn 0.22.2 Feature selection and performance evaluation skrebate 0.6 For SURF based feature selection imblearn 0.6.2 Implements a balanced random forest classifier
Illustration of cfRNA-SEEK work flow